Diese Seite ist nicht mit dem Internet Explorer kompatibel.
Aus Sicherheitsgründen empfehlen wir Ihnen, einen aktuellen Browser zu verwenden, z. B. Microsoft Edge, Google Chrome, Safari oder Mozilla Firefox.
Simulation molekulare Diffusion
mit VGSTUDIO MAX
Simulieren Sie die molekulare Diffusion auf CT-Scans unterschiedlicher Werkstoffe mit dem Modul Transportphänomene-Simulation für VGSTUDIO MAX.
Simulation der molekularen Diffusion
Die Diffusion beschreibt den Fluss von Molekülen von einem Bereich mit höherer Konzentration in einen Bereich mit niedrigerer Konzentration.
Das Modul Transportphänomene-Simulation für VGSTUDIO MAX
- berechnet die stationäre molekulare Konzentration und den Fluss, die asymptotisch erreicht werden, wenn eine Probe mit Reservoirs mit unterschiedlichen molaren Konzentrationen verbunden ist, wobei angenommen wird, dass die Poren der Probe vollständig mit dem Lösungsmittel gefüllt sind und die Konzentration in den Reservoirs konstant ist, wie in sehr großen Reservoirs;
- arbeitet direkt auf den Voxeldaten und verwendet die subvoxelgenaue, lokal adaptive Oberflächenbestimmung in VGSTUDIO MAX;
- verfügt über einen „Experimentmodus“ zur Durchführung eines virtuellen Experiments zur Diffusion von Molekülen sowie über einen „Tensormodus“ zur Berechnung des Tensors der molekularen Diffusion.
Die Berechnung der molekularen Diffusion basiert auf den folgenden Differenzialgleichungen für die stationären Konzentrations‑ und molaren Flussdichte-Felder in einem Zweikomponentenwerkstoff:

wobei Ω der gesamte Simulationsbereich und Ωₐ der Komponentenbereich a (mit a = 1, 2) ist. Es wird angenommen, dass sich Ω₁ und Ω₂ nicht überlappen und ihre Vereinigungsmenge gleich Ω ist. C ist die molekulare Konzentration, J die molare Flussdichte, Dₐ der Diffusionskoeffizient der Komponente a, Δ der Laplace-Operator und grad der Gradient-Operator.
Experimentmodus
Im Experimentmodus führt die Software ein virtuelles Experiment auf den CT-Daten einer Struktur durch, wobei der Molekültransport durch die Struktur von einer Einlassebene zur einer parallel dazu verlaufenden Auslassebene simuliert wird. Als Randbedingungen können die Optionen „versiegelt“ oder „eingebettet“ senkrecht zur Einlass- und Auslassebene ausgewählt werden. Als treibende Kraft für den Fluss dient ein Konzentrationsunterschied.
Berechnungsergebnisse
Die Ergebnisse werden als Visualisierung der molaren Flussdichte und der Stoffmengenkonzentration in 2D und in 3D dargestellt. Die Richtung der molekularen Diffusion kann über Stromlinien in 2D und 3D visualisiert werden:
- molare Flussdichte
- Stoffmengenkonzentration
- Stromlinien der molekularen Diffusion
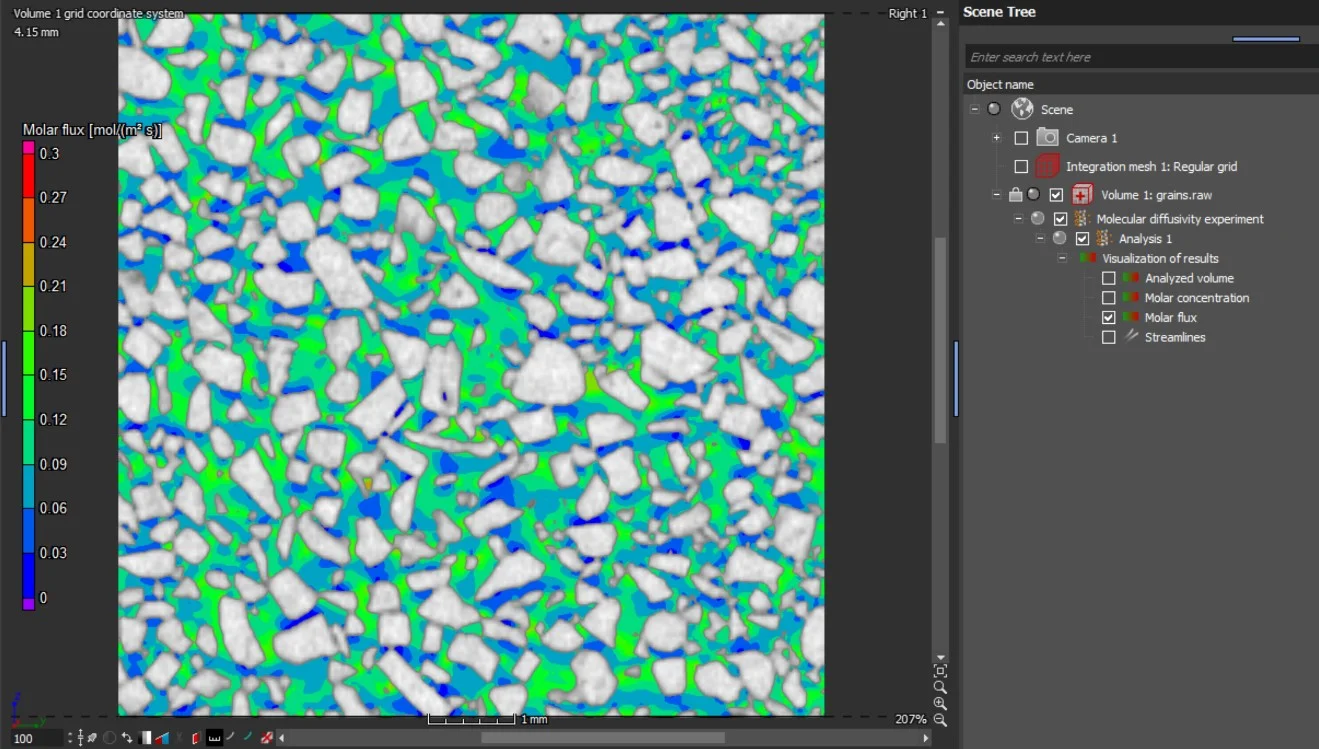
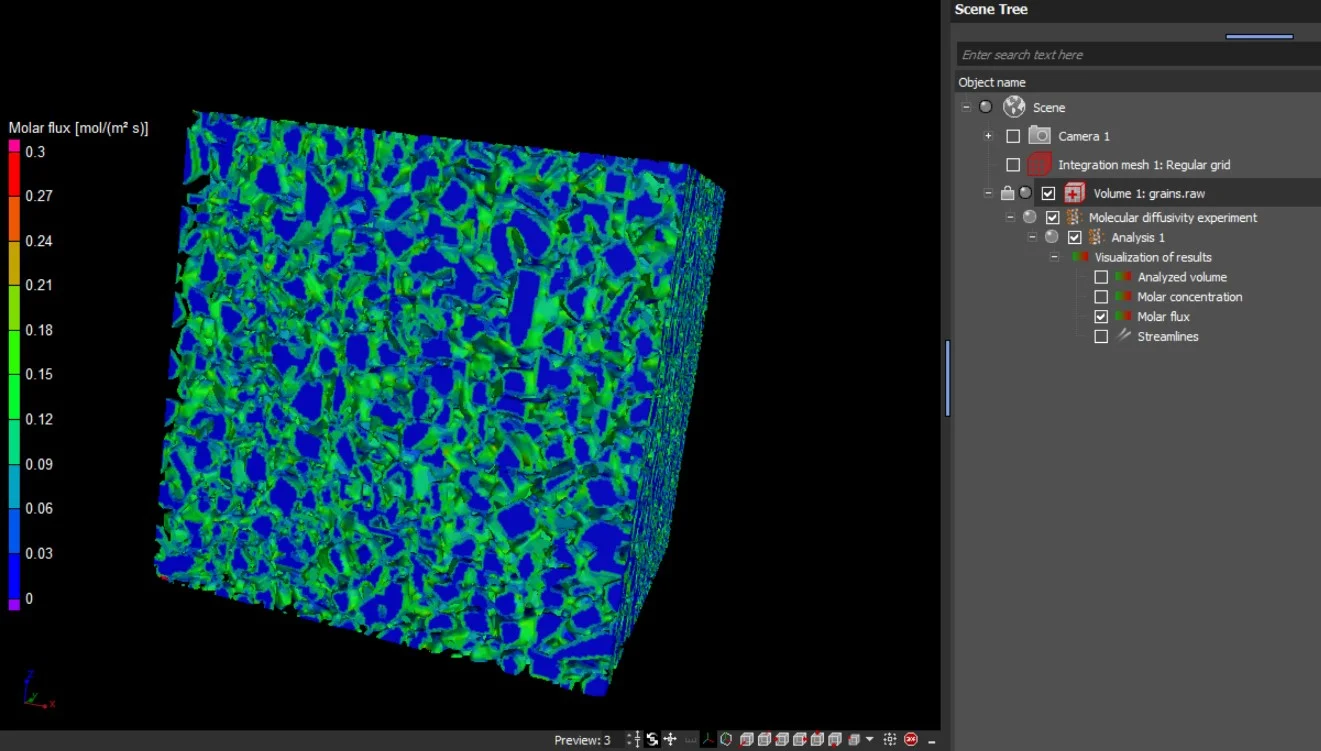

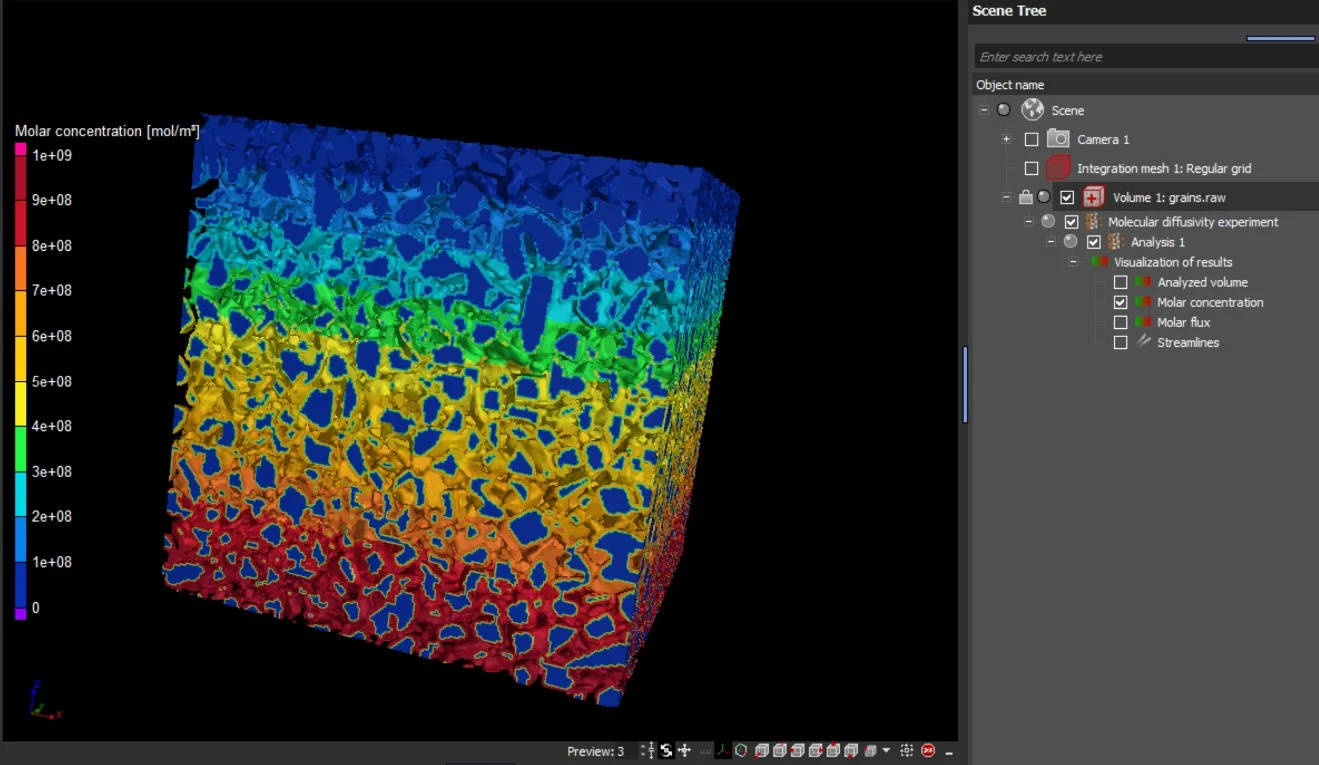
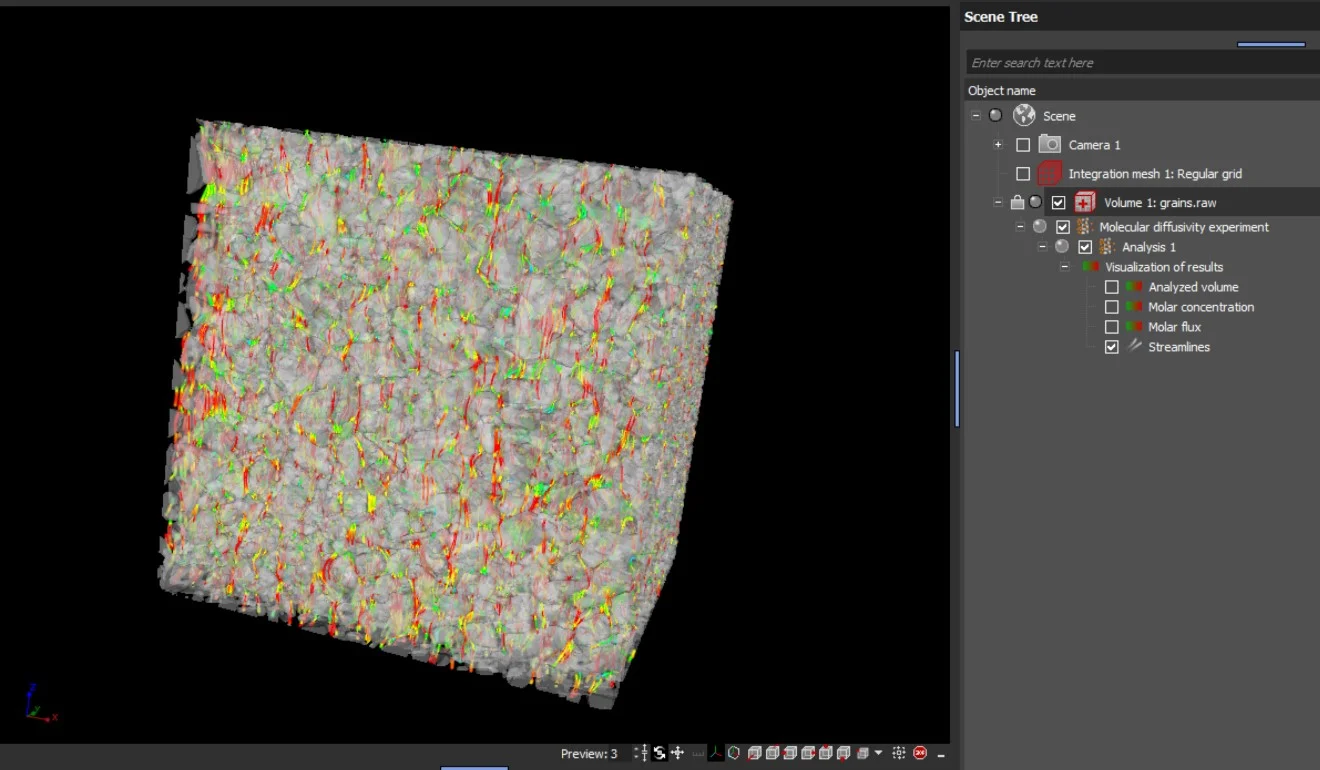
Weitere Ergebnisse
Zusätzlich werden folgende Ergebnisse tabellarisch aufgelistet:
- effektiver Diffusionskoeffizient
- diffusive Tortuosität (geometrisch und algebraisch)
- Formationsfaktor
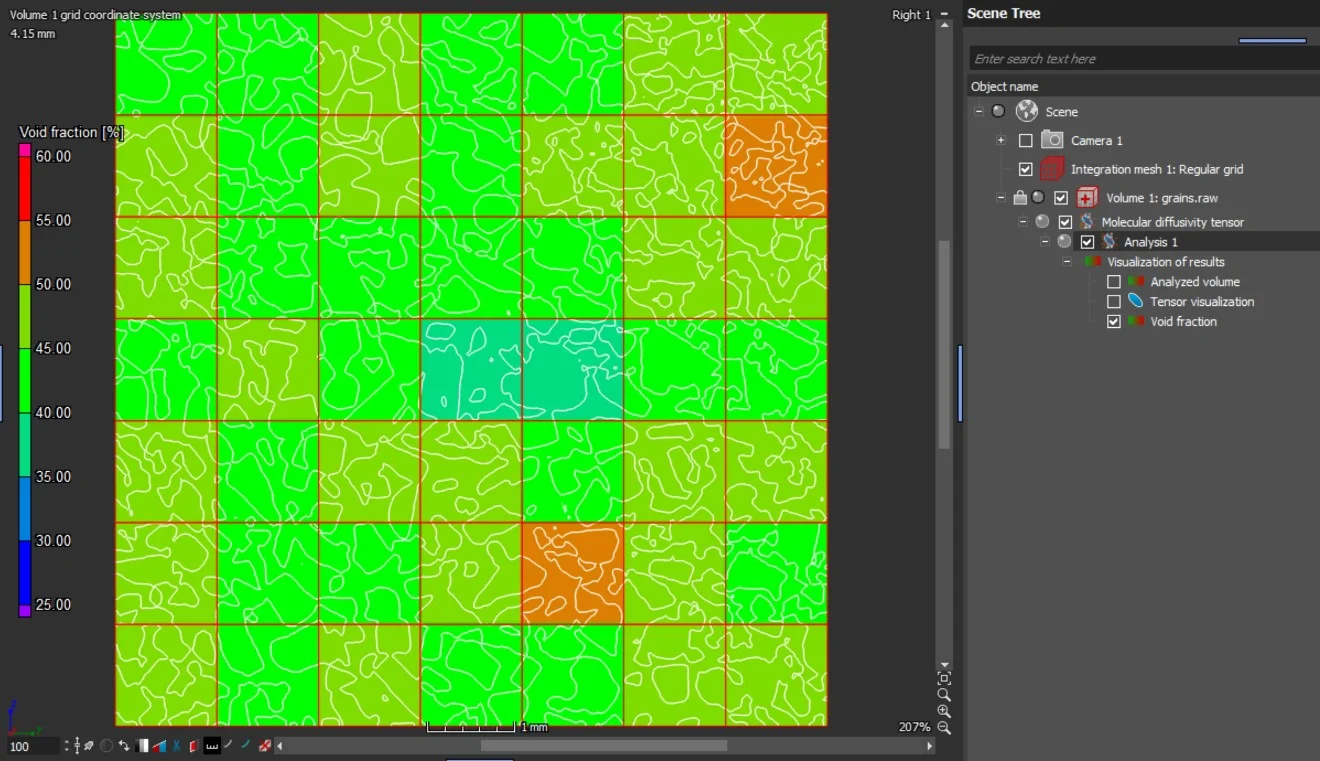
- Hohlraumanteil
- Gesamtmassentransport
- Konzentrationsgradient
Die folgenden Ergebnisse werden als Kurvendiagramme in Fließrichtung dargestellt:
- molare Flussdichte
- Hohlraumanteil
- Gesamtmassentransport
Tensormodus
Im Tensormodus berechnet die Software die Komponenten des Tensors der molekularen Diffusion. Die Berechnung des Tensors der molekularen Diffusion kann auf der gesamten Struktur erfolgen oder – anhand eines Integrationsnetzes – auf der gerasterten Struktur.
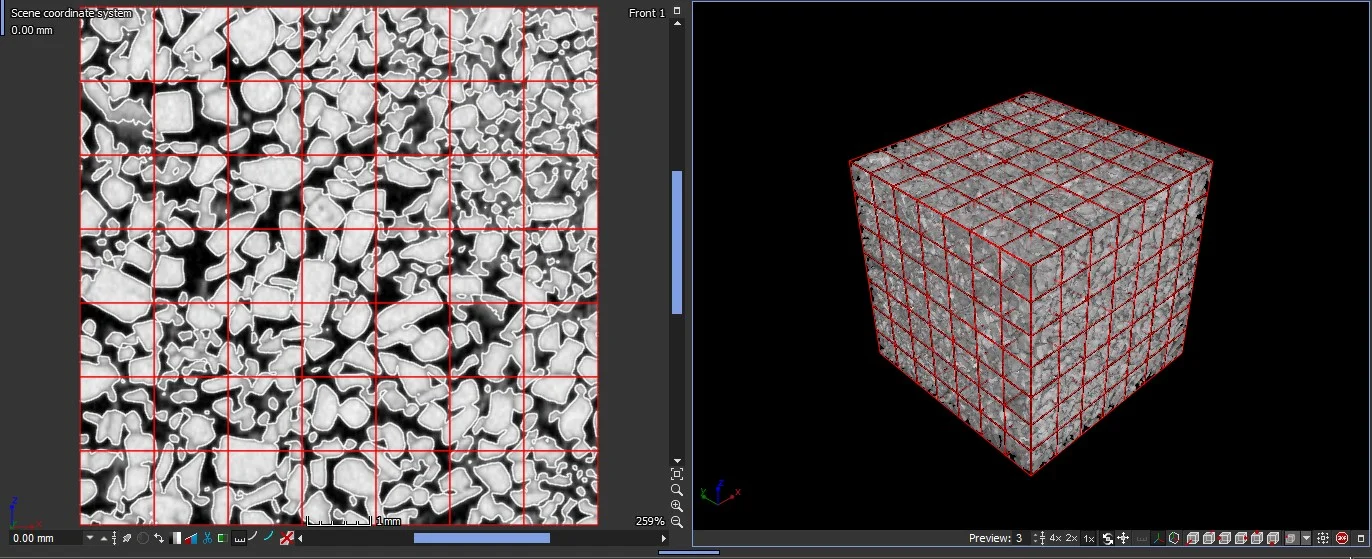
Ergebnisse
Die Software stellt die Ergebnisse der Berechnung des Tensors der molekularen Diffusion als Visualisierung in 2D und in 3D dar.
- Die Ergebnisse werden als Ellipsoid im Zentrum des simulierten Volumens dargestellt.
- Wenn die Analyse auf einem Integrationsnetz durchgeführt wurde, erfolgt die Darstellung der Ergebnisse als Ellipsoid im Zentrum jeder Zelle.
- Die Hauptachsen des Ellipsoids sind an den Eigenvektoren des Tensors ausgerichtet, während die Länge der Achsen proportional zu den Eigenwerten des Tensors ist.
- Wenn die Analyse auf einem Integrationsnetz durchgeführt wird, können außerdem der Tensor der mittleren effektiven molekularen Diffusion und der Hohlraumanteil sowie die Eigenwerte des Tensors der molekularen Diffusion in 2D und in 3D dargestellt werden.

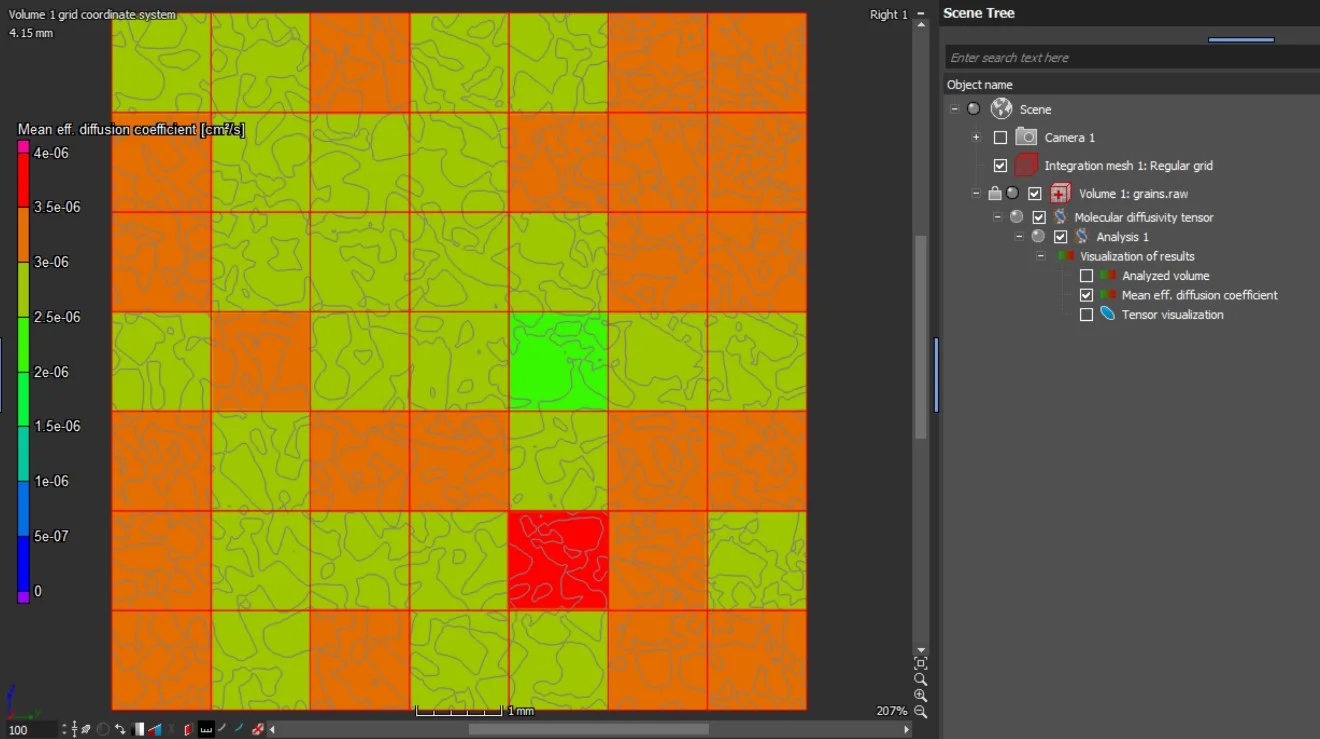


Zusätzlich zu den Eigenwerten und Eigenvektoren des Tensors werden die Tensorkomponenten des effektiven Diffusionskoeffizienten in Bezug auf das Simulationskoordinatensystem in einer Tabelle aufgelistet.
Vorteile
Benutzerfreundlich
- Keine Simulationserfahrung erforderlich
- Keine Vernetzung erforderlich
Effizient
- Nahtlose Arbeitsabläufe in einer einzigen Software – von der Materialsegmentierung bis zur Simulation
Wirklichkeitsgetreu
- Alle mikrostrukturellen Details werden durch die subvoxelgenaue Oberflächenbestimmung erfasst